Molecular Solar Thermal Systems:
In the recent decade, a growing attention was devoted to find alternative solutions to the problem of energy storage. Especially, solar energy is one of the most available and clean sources of energy, but its storage is still an open question. A possible solution could be given by MOlecular Solar Thermal (MOST) systems: a molecule could be designed as a low-energy isomer that, after activation by solar light, can be transformed into a high-energy isomer. The difference in energy between the two isomers constitutes the stored energy (Estored). Apart from high photon absorption intensity and efficiency to initiate the photoisomerization process, other criteria need to be concomitantly fulfilled to obtain a suitable MOST system:
– The high-energy isomer has to be photochemically stable at room temperature.
– The ground state energy barrier (i.e., the activation energy, Ea) to release the stored energy has to be high enough to guarantee chemical stability of the high-energy isomer, and at the same time, it should be conveniently lowered by the addition of a proper catalyst to release Estored at demand.
– To be of technological interest, the storage density (Dstorage = ΔHstorage/M, where ΔHstorage is the difference in enthalpy between high- and low-energy isomers and M is the molecular weight) needs to be maximized.
– For device purposes, the overall MOST system (i.e., both isomers) should be liquid to ensure a flux among different device compartments.
Therefore, it can be realized that, although simple in its chemical concept, the optimization of a MOST system is far from being a trivial task. For these reasons, we have studied MOST systems, aimed both at understanding the properties of the well-known and largely applied norbornadiene/quadricyclane (NBD/QC) system or to propose novel MOST systems.
MOST systems based on NBD/QC
Studying in detail the reactivity of a given system is crucial to propose modifications aimed at enhancing its properties. For instance, we have studied in detail the NBD↔QC reactivity considering low-lying valence and Rydberg excited states. We tackled this challenge by performing extensive multireference perturbation theory calculations (CASPT2) to properly include static and dynamic electronic correlation. In this case we show the relevance of including the dynamic electron correlation to avoid a misleading description of the photochemical pathways as it affects the energetic order of the excited states and, as a consequence, the overall description of the relaxation channels.

Figure adapted from DOI: 10.1002/cptc.202200214
Novel MOST systems
In the last years, we have proposed novel MOST systems as alternatives to NBD/QC which is nowadays the most relevant building block to design MOSTs. In this regard, we have followed two approaches: i) conversion of a molecular photoswitch into a MOST and ii) study of novel systems.
Regarding the first approach, we have proposed design principles to convert the well-known photoswitch azobenzene into a MOST system. Indeed, although in principle any photoswitch could be a MOST, the many criteria to be fulfilled for MOST efficiency have hampered the use of photoswitches in this field. For instance, the thermal stability of the high-energy isomer is usually not fulfilled by photoswitches. This inconvenience can be solved by designing modifications decreasing the energy of the high-energy isomer or increasing the transition state energy. However, these modifications should not compromise other MOST properties such as a large Estored or the absorption window of the low-energy isomer. Another common inconvenience is the undesired eventual absorption of solar light by the high-energy isomer.

Figure adapted from DOI: 10.1002/adsu.202200097
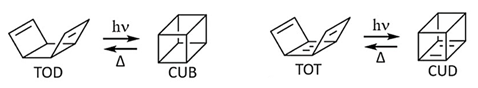
The second approach is completely innovative, aiming at discovering novel molecular systems. For instance, we have proposed a MOST system based on tricyclooctadiene (TOD)↔cubane (CUB) or tricyclooctatetraene (TOT)↔cubadiene (CUD). To get insight into their suitability as MOST systems, we have studied computationally their reactivity through multi-configurational quantum chemistry methods. We have shown the electronic absorption properties, photochemical, and ground-state mechanistic paths of both systems, highlighting their pros and cons in the quest for novel MOST systems.
Presently, different reactivities other than [2+2] photocycloaddition are under study.