Proteins’ functional response:
In recent years, we have studied proteins with the goals of obtaining structural, thermodynamical and kinetical data to propose a certain function. Responding to the emergence caused by SARS-CoV-2, we have investigated various aspects of some proteins involved in this disease, including their structure, mechanisms of action, and inhibition. Presently, we are applying the same methods and updated ones to gain such atomistic-resolution information.
Structure
Regarding the structural analysis of proteins, we have studied for instance the differences in the ACE2/RBD interaction depending on the SARS-CoV-2 variant (wild-type, alpha and delta), showing different mutations in the Spike protein. We have shown that extensive all-atom molecular dynamics simulations, combined with machine learning-driven analysis (as a collaboration with CNRS at Nancy, France), can comparatively indicate how these mutations alter the molecular interaction network in comparison to the wild-type. For instance, it was found that hydrophilic and hydrophobic interactions affect both the structure of the ACE2/RBD interface and their formation/disruption dynamics. This methodology can be easily applied to other protein complexes or biomolecular systems.
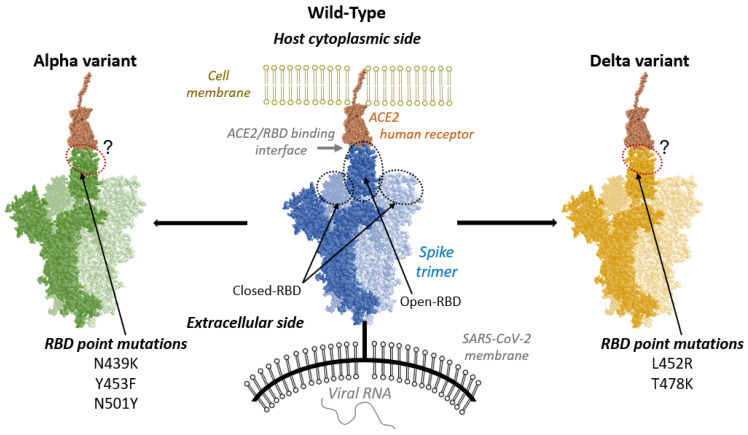
Figure adapted from DOI: 10.3390/ijms24032517
Our approach can be applied indistinctly to globular and transmembrane proteins. Indeed, we have reported the first detailed structural analysis of Nsp6, a transmembrane protein of interest for SARS-CoV-2, mapping the protein/membrane physicochemical interactions to investigate the structural variables anchoring the protein within the lipid bilayer.
Function
To propose structure-to-function relationships, we have performed all-atom equilibrium and enhanced sampling molecular dynamics simulations. In particular, we have studied the interaction between the SARS Unique Domain and RNA guanine quadruplexes, playing a crucial role in evading the host’s immune response, thereby facilitating viral infection of human cells. Applying this methodology, the role of different binding modes as well as their stability can be retrieved. Moreover, by performing free energy calculations it can be concluded which binding mode is thermodynamically preferred.

Figure adapted from DOI: 10.1021/acs.jpclett.0c01097
Another example of applying enhanced sampling molecular dynamics simulations to get information about a protein function is the study of the viral 3′ Open Reading Frame (ORF3a) of SARS-CoV-2: we have shown, again in collaboration with CNRS at Nancy, France, that ORF3a, initially embedded in a model lipid bilayer, can form a channel across the membrane, facilitating ion transport from extra- to intra-cellular media and vice versa. Moreover, by performing free energy calculations we have facilitated the free energy profile for both K⁺ and Cl⁻ ion transfer from the cytosol to the extracellular space.

Figure adapted from DOI: 10.1021/acs.jpclett.2c01102
Mechanisms of inhibition
The function of a given protein can be inhibited using ligands leading to covalent or non-covalent inhibition. The study of these mechanisms is relevant not only for the understanding of the process but also to propose novel ligands acting as inhibitors. We have studied from a molecular level these two types of inhibition, showing the relevant information that can be retrieved. In particular, we have analyzed: i) non-covalent inhibition of human and viral proteins involved in the infection cycle of SARS-CoV-2 by drug interaction, and ii) covalent inhibition of SARS-CoV-2 Papain-like Protease.
i) Non-covalent interactions: Combining molecular docking and molecular dynamic simulations, we have analyzed the effect of well-known drugs on disrupting SARS-CoV-2 recognition by inhibiting its interaction with ACE2. Comparing the drugs interactions with ACE2 let us conclude which drug could lead to a stronger interaction, resulting in a potential inhibitor of the protein under study. Moreover, instead of studying diverse drugs as inhibitors for a given protein, we have also evaluated a given drug (ivermectin) that could act as an inhibitor for diverse proteins. This way, we can actively help in the study of multi-purposing drugs, rationalizing which targets will be most likely inhibited.

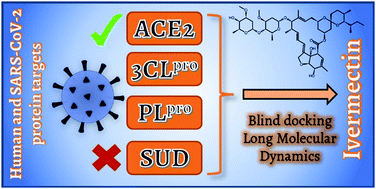
Figures adapted from DOI: 10.1021/acs.jpclett.0c02203 and 10.1039/D1CP02967C
ii) Covalent inhibition: Another approach for protein inhibition is the use of a ligand modifying covalently its active site, i.e., forming a covalent bond between ligand and protein. Understanding the mechanism of this process could aid in the design of more efficient and selective inhibitors, improving therapeutic strategies. For instance, we have computationally studied the covalent inhibition of the Papain-like Protease (PLpro) of SARS-CoV-2 by addition of the VIR251 ligand. To investigate this inhibition mechanism, we employed classical molecular dynamics simulations and quantum mechanics/molecular mechanics (QM/MM) calculations to evaluate the energetic landscape and structural changes at each step of the reaction mechanism, elucidating its thermodynamics and kinetics. This methodology could be applied to whatever covalent inhibition reaction of any functional protein.
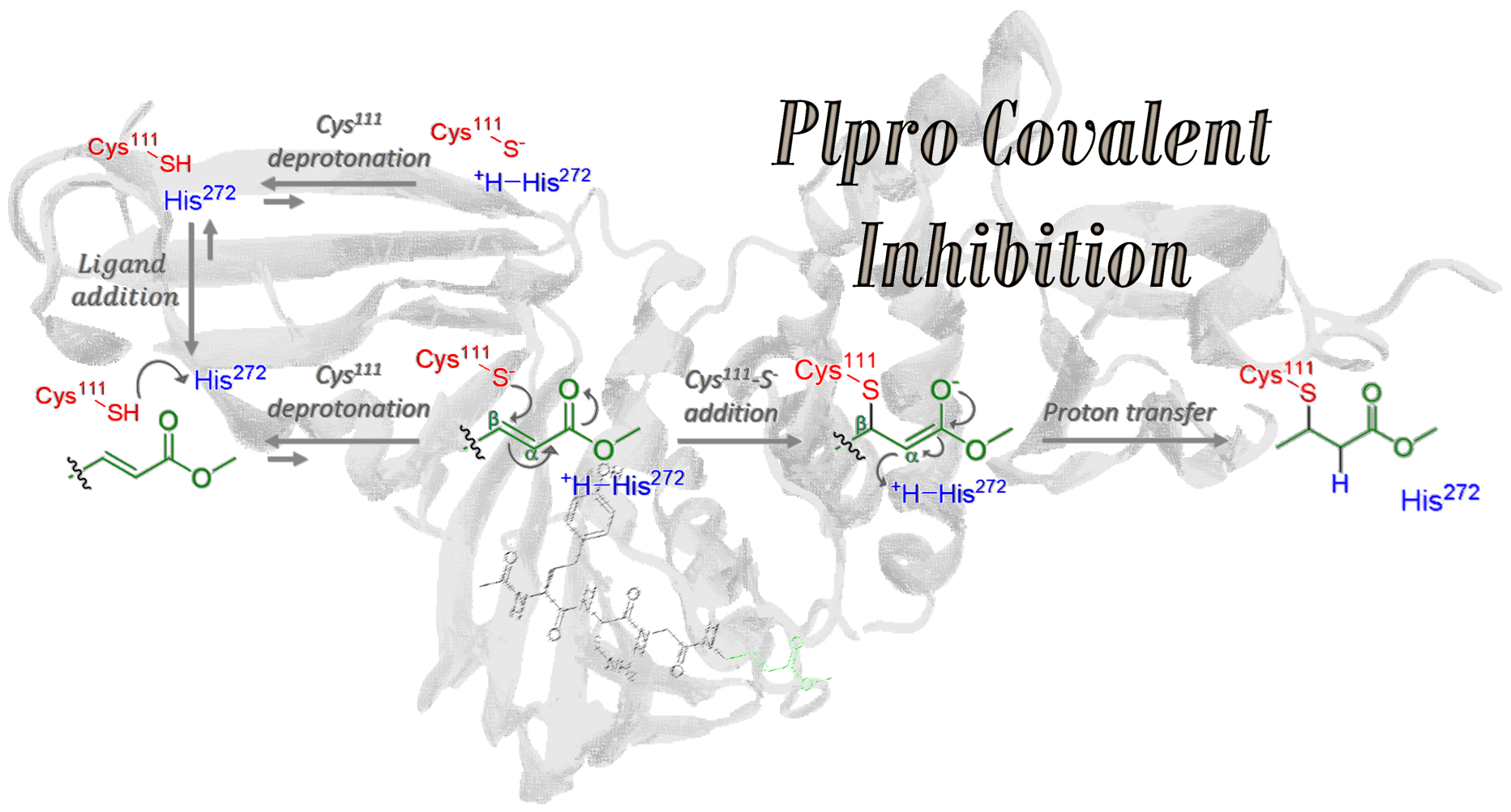
Figure adapted from DOI: 10.3390/ijms23105855